TP 3. Predicción Estructural (A)
Recursos Online
- HHPred: https://toolkit.tuebingen.mpg.de/tools/hhpred
- UCLA-DOE LAB: https://saves.mbi.ucla.edu/
- MolProbity: http://molprobity.biochem.duke.edu/
- AlphaFold Server: https://alphafoldserver.com/
Materiales
Objetivos
- Familiarizarse con las técnicas de modelado por homología y predicción estructural
- Analizar la calidad de los modelos obtenidos
Parte 1 - Modelado por Homología
Ejercicio 1. Modelado de la Proteína COX17.
La proteína citocromo c oxidasa es un componente terminal de la cadena respiratoria mitocondrial catalizando la transferencia de electrones al oxígeno.
Es un complejo formado por tres subunidades catalíticas codificadas por genes mitocondriales y nucleares.
En humanos, la metalochaperona de cobre citocromo c oxidasa está codificada por el gen COX17, está involucrada en el reclutamiento de cobre a la mitocondria y en el ensamblado del complejo que forma parte de la cadena respiratoria. La proteína humana comparte un 92% de identidad de secuencia con las proteínas homólogas de ratón y rata.
Si bien se conoce su función, la mecánica de la reacción y las interacciones establecidas en el complejo aún no se conocen en profundidad.
Estudios recientes revelaron que la acumulación de cobre en la mitocondria conduce a la disfunción mitocondrial, apoptosis celular y fibrosis renal, por disrupción de la actividad de la citocromo c oxidasa.
En riñones fibróticos, el gen COX17 se encuentra sobre-expresado.
En ratones, el knockdown de COX17 produce un aumento en la acumulación de cobre mitocondrial, inhibición del complejo c oxidasa, aumento de la disfunción mitocondrial, apoptosis celular y fibrosis renal, mientras que la sobre-expresión permite la descarga del cobre mitocondrial protegiendo la función y aliviando la fibrosis renal.
En este ejercicio modelaremos la estructura de la chaperona de cobre de levaduras (Q12287).
-
Ingrese la secuencia de la proteína COX17 de levaduras en HHPred
2. Haga click en Submit en la parte inferior de la página y seleccione el hit que le parezca más conveniente:>sp|Q12287|COX17_YEAST Cytochrome c oxidase copper chaperone OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) OX=559292 GN=COX17 PE=1 SV=3 MTETDKKQEQENHAECEDKPKPCCVCKPEKEERDTCILFNGQDSEKCKEFIEKYKECMKG YGFEVPSAN- ¿por qué es el más conveniente?
- ¿Que e-value tiene?
- ¿que porcentaje de identidad posee con su proteína (en la pate inferior está el alineamiento)?
Luego seleccione en la parte superior Model using selection.
- ¿Qué se muestra en la nueva ventana? (Mueva la barra inferior para ver que hay en la ventana).
-
Haga click en Forward to Modeller y luego en Submit.
Atencion
De ser necesario ingrese la siguiente key: MODELIRANJE en el recuadro que dice Modeller key y luego haga click en Submit.
- ¿Qué aparece en la nueva ventana?
-
Descargue el archivo PDB (Download PDB File)
-
La herramienta MolProbity permite analizar características esperadas en un modelo de buena calidad.
-
Ingrese el modelo obtenido en el paso anterior donde dice Choose file y luego haga click en Upload >
-
En la nueva página haga click en Continue >
-
Luego haga click en Analyze geometry without all-atom contacts
-
En la siguiente página asegúrese que las siguientes opciones están seleccionadas:
En la sección Charts, plots, and tables dentro de Universal
-
Clashes & clashscore
-
Geometry evaluation
-
Protein
-
Ramachandran plots
-
Rotamer evaluation
-
Cβ deviations
-
Cis-Peptide evaluation
-
Show cis-nonPro and twisted peptide statistics even if the model has none
-
CaBLAM backbone evaluation
Por último en Other options
-
Create html version of multi-chart
-
List all residues in multi-chart, not just outliers
-
Remove residue rows with ' ' altloc when other alternate(s) present
-
-
Luego, haga click en Run programs to perform these analyses >
-
Observe la tabla y responda:
-
¿Qué métricas se están evaluando?
-
Investigue el gráfico de ramachadran (debe descargar el PDF)... ¿Puede detectar los outliers?
-
Vaya a Multi-criterion visualizations y haga click en View
-
¿Qué residuos se encuentran en zonas menos favorecidas en el ramachandran?
-
En base a los resultados ¿Considera que su modelo es de buena calidad?
-
CaBLAM (C \(\alpha\) Based Low-resolution Annotation Method)
Es un criterio en MolProbity para estructuras de baja resolución (>2 Å) que decta errores en el backbone
-
-
La herramienta Verify3D permite determinar la compatibilidad de un modelo 3D de una proteína con su secuencia aminoacídica en base a cuál es el ambiente en el cual se encuentra cada residuo y la compatibilidad con la estructura secundaria en la que se encuentra.
Vaya a la web de UCLA-DOE LAB, suba el archivo PDB obtenido en Choose file y clickee en Run programs.
- Seleccione Verify3D y espere por los resultados. Haga click en Results.
El gráfico reporta la calidad del modelo por posición y en él se observan tres regiones:
Info
- Posiciones con score menor a cero están mal modeladas,
- Posiciones con score entre cero y 0.1 están pobremente modeladas,
- Posiciones con score mayor a 0.1 están modeladas con buena calidad.
Verify 3D asigna como aceptado a un modelo con más del 80% de las posiciones posiciones con un score promedio en el área bien modelada.
Observe el resultado obtenido (Si tarda haga click en el botón Check status) y responda:
- ¿Cuál es el porcentaje de residuos con un score promedio en el área de bien modelados?
- ¿Qué región está pobremente modelada según Verify 3D?
-
La herramienta Procheck permite analizar la calidad de la geometría de los residuos en una estructura proteica dada en comparación a parámetros estereoquímicos derivados de estructuras tridimensionales de alta resolución ya conocidas.
En la parte superior de la página de los resultados de Verify 3D vaya a Control Panel
Seleccione Procheck y espere por los resultados.
-
Investigue el Ramachandran Plot. Reconozca las regiones a los distintos elementos de estructura secundaria y responda:
- ¿Cuántas estructuras se utilizaron para construir este Ramachandran?
- ¿Qué residuos no están en el área esperada?
- ¿Qué criterio se utiliza para considerar que el modelo es de buena calidad?
- ¿Qué porcentaje de residuos en la estructura modelada se encuentran en las regiones más favorecidas?
-
Investigue los gráficos de ramchandran para todos los residuos en All Ramachandrans debe elegir el pdf.
- ¿Cuántas estructuras se utilizaron para construir este Ramachandran?
- ¿Qué residuos no están en el área esperada?
-
Investigue los gráficos de las longitudes de enlace en la cadena principal (M/c bond lengths) y los ángulos de unión de la cadena principal (M/c bond angles).
- ¿Existen aminoácidos que se alejen significativamente de los resultados esperados?
-
-
En base a los resultados obtenidos por Verify 3D y ProCheck responda: ¿Es bueno el modelo? ¿Por qué?
-
Abra chimera y cargue su modelo:
Si no funciona, el pdb se encuentra en su carpeta de datos y puede utilizar:
File → Open
-
Un paso importante para asegurar la calidad del modelo es comparar el modelo obtenido contra el templado utilizado
-
¿Qué templado utilizó? Importante: Identifique el PDB y la cadena
-
Abra la estructura en chimera, en la misma ventana donde ya tiene su modelo.
-
-
Para tener una noción de cuán similar es la estructura de dos proteínas, podemos realizar un Alineamiento Estructural, que consiste en superponer las estructuras de ambas proteínas en el espacio intentando alinear sus cadenas aminoacídicas. Para alinear estructuras en chimera se utililiza el Matchmaker.
Vaya a Tools → Structure Comparison → MatchMaker
Se abrirá una nueva ventana.
-
En Reference structure (el panel de la izquierda) puede seleccionar una de las estructuras de referencia. Esta estructura es la que se mantendrá fija. (Ej. El modelo obtenido)
-
En Structure(s) to match (el panel de la derecha) seleccione la estructura que será superpuesta y alineada con la que se eligió como referencia. (Ej. el templado)
-
En Matching asegurése que Iterate by pruning long atom pairs untilo no pair exceeds está clickeado.
O bien, ingrese en la command line:
mm #0 #1- Observe el resultado del alineamiento: ¿Son parecidas las estructuras? ¿En donde se observan las mayores diferencias?
Vaya a Favorites → Reply Log
- ¿Cuál es el RMSD reportado?
-
-
Para ver cómo se corresponde el grado de similitud estructural con el grado de similitud en secuencia podemos realizar un alineamiento de ambas secuencias guiado por el alineamiento estructural. Para esto, vaya a:
Tools → Structure comparison → “Match->Align”
Ahora, observando la estructura y el alineamiento responda:
I. ¿Qué son las regiones marcadas en rosa en el alineamiento?
II. ¿Este alineamiento, identifica regiones que no alinean estructuralmente? ¿A qué se debe?
III. En la parte superior de la ventana del alineamiento de secuencia vaya a Headers y seleccione RMSD:ca
- ¿Qué regiones poseen mayor RMSD? ¿A qué elementos estructurales corresponden? Para responder esto, seleccione estas regiones con el mouse en el alineamiento y visualícelas en la estructura alineada.
-
Para cuantificar el alineamiento de secuencia obtenido, podemos calcular el % de identidad de secuencia. Para ello, en la ventana del alineamiento de secuencias vaya a:
Info → Percent identity.
Seleccione una estructura en Compare y la otra estructura en with. En Divide by seleccione longer sequence length. Presiona en Ok.
-
¿Qué valor de identidad de secuencia obtiene? ¿Porque cree que difiere del reportado anteriormente?
-
En la parte superior de la ventana del alineamiento de secuencia vaya a Headers y seleccione Conservation ¿Las sustituciones observadas en las secuencias son conservativas?
-
-
En base a los resultados obtenidos. ¿Intentaría obtener experimentalmente la estructura de la proteína, o confiaría en el modelo?
Parte 2 - AlphaFold
AlphaFold
En los últimos años, hubo un crecimiento continuo en el número de estructuras de proteínas determinadas experimentalmente depositadas en el PDB (actualmente >250.000). Esto, junto con la explosión de la secuenciación (millones de secuencias) y el desarrollo de técnicas de deep learning benefició el desarrollo de algoritmos de predicción de estructura tridimensional de proteínas.
Hasta el 2021, los algoritmos de predicción de estructura tridimensional de proteínas se basan en dos aspectos complementarios: las interacciones físicas (o contactos) o la historia evolutiva de la proteína (homología). Sin embargo, y a pesar de los avances, la mayoría de los algoritmos de predicción no son muy precisos si se carece de un homólogo cercano con una estructura tridimensional resuelta experimentalmente.
A partir de la secuencia primaria de una proteína (Fig 1), AlphaFold utiliza una red neuronal para la predicción estructural de alta precisión (en la mayoría de los casos), la cual aumenta con el uso de estructuras homólogas. AlphaFold puede incluso predecir con alta precisión las cadenas laterales si el backbone es preciso.
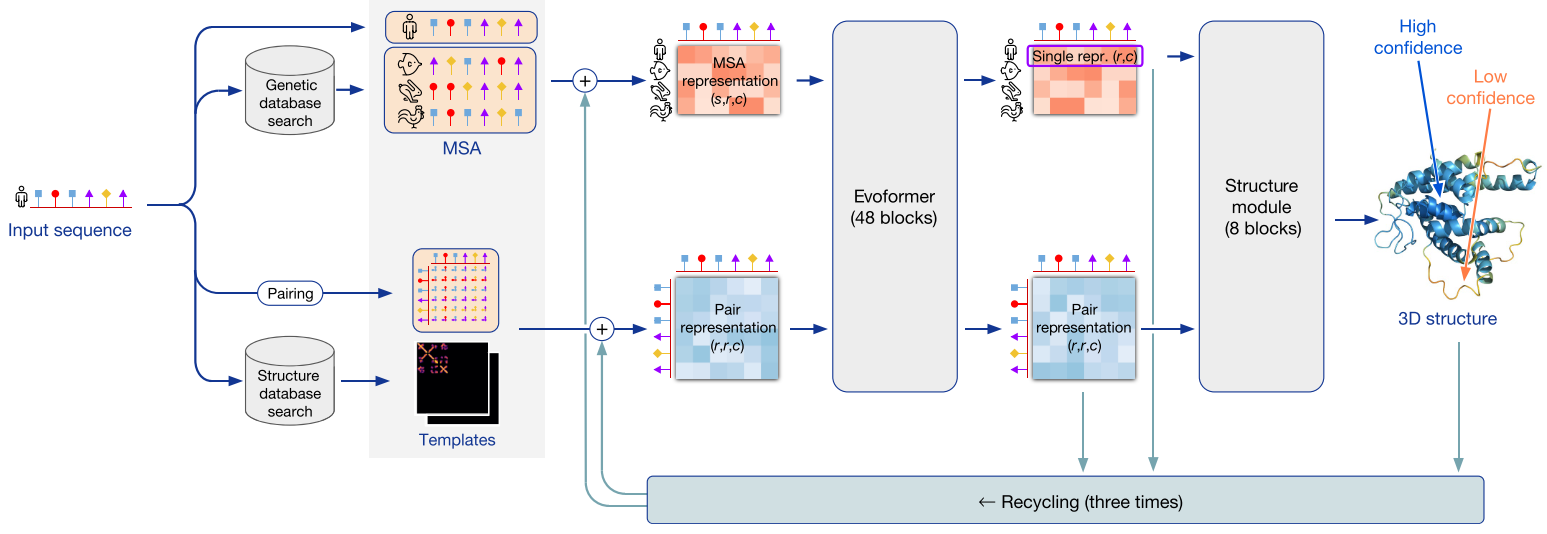
Arquitectura
AlphaFold3 utiliza una arquitectura de red similar a la de AlphaFold2, que utiliza como inputs el alineamiento de secuencias (MSAs) y una representación de todos los pares de residuos de la secuencia que luego es utilizado por un módulo estructural para generar la estructura. AlphaFold2 utiliza un algoritmo iterativo que se basa en la arquitectura Evoformer para procesar los inputs y junto con el módulo estructural generar una representación tridimensional de la proteína query. AlphaFold3 para reducir la cantidad del procesamiento del MSA, reemplaza el algoritmo Evoformer con el módulo Pairformer que utiliza un bloque más pequeño de embedding para el MSA. Algunas de las mejoras en la performance en la predicción están directamente relacionadas con la reducción en la dependencia de la señal del MSA. De todas maneras, para la predicción de estructura de proteínas, AlphaFold3 se basa fuertemente al igual que AlphaFold2 en la co-evolución de residuos observada en el MSA.
A diferencia de AlphaFold2 que predice las posiciones de aminoácidos y sus cadenas laterales, AlphaFold3 predice las coordenadas de los átomos individuales en el complejo, lo cual facilita la predicción de distintos tipos de moléculas.
Tanto AlphaFold2 como AlphaFold3 dividen al complejo en tokens. En AlphaFold2 los tokens corresponden directamente a aminoácidos. En AlphaFold3, el enfoque de un token por átomo para toda la estructura, permitiría una mayor flexibilidad para modelar distintas moléculas, sin embargo, pone en compromiso restricciones de memoria. Por lo tanto, intenta una estrategia que permita cierto equilibrio. Un token puede ser un aminoácido, un nucléotido, un átomo de un ligando, un ión o por ejemplo, un átomo de un aminoácido o nucleótido modificado químicamente.
Por ejemplo, una estructura de 100 aminoácidos y un ligando de 20 átomos, requiere 120 tokens.
A diferencia de AlphaFold2, las métricas de confianza son calculadas por tokens y no por aminoácidos.
Mientras AlphaFold2 es un modelo no-generativo que identifica patrones en los datos. AlphaFold3 usa un modelo generativo de machine learning. Los modelos generativos crean nuevos datos similares a los ejemplos que aprendieron.
Métricas de confianza
AlphaFold incorpora métricas de confianza de la predicción.
-
La principal métrica de confianza es el test pLDDT (predicted local-distance difference test) el cual es un predictor confiable del test de diferencias en las distancias Cα (LDDT-Cα) y evalúa principalmente la correctitud del modelo a nivel local (estimando el error en distancias de un Cα con Cα vecinos en un rango de 15Å).
-
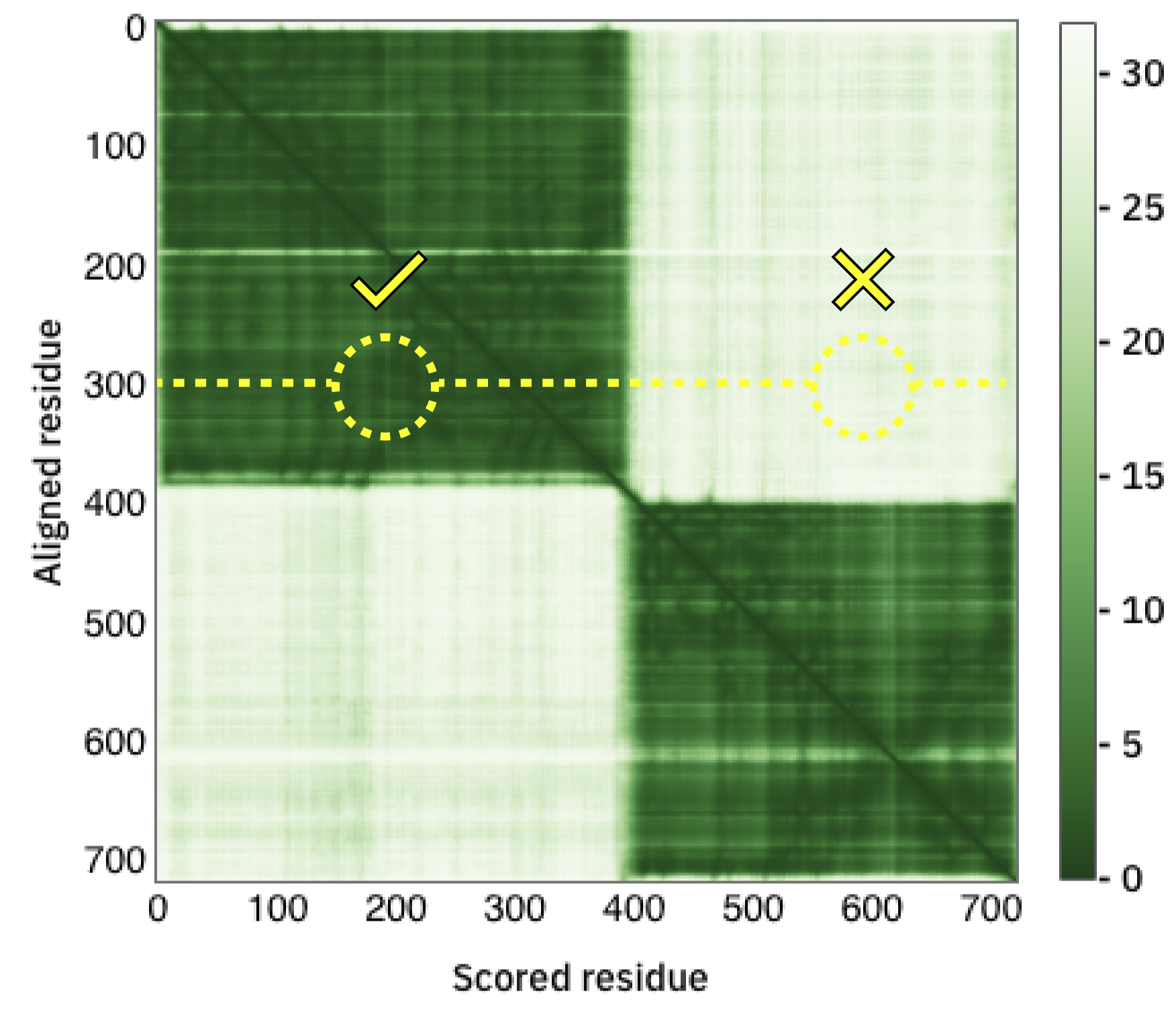
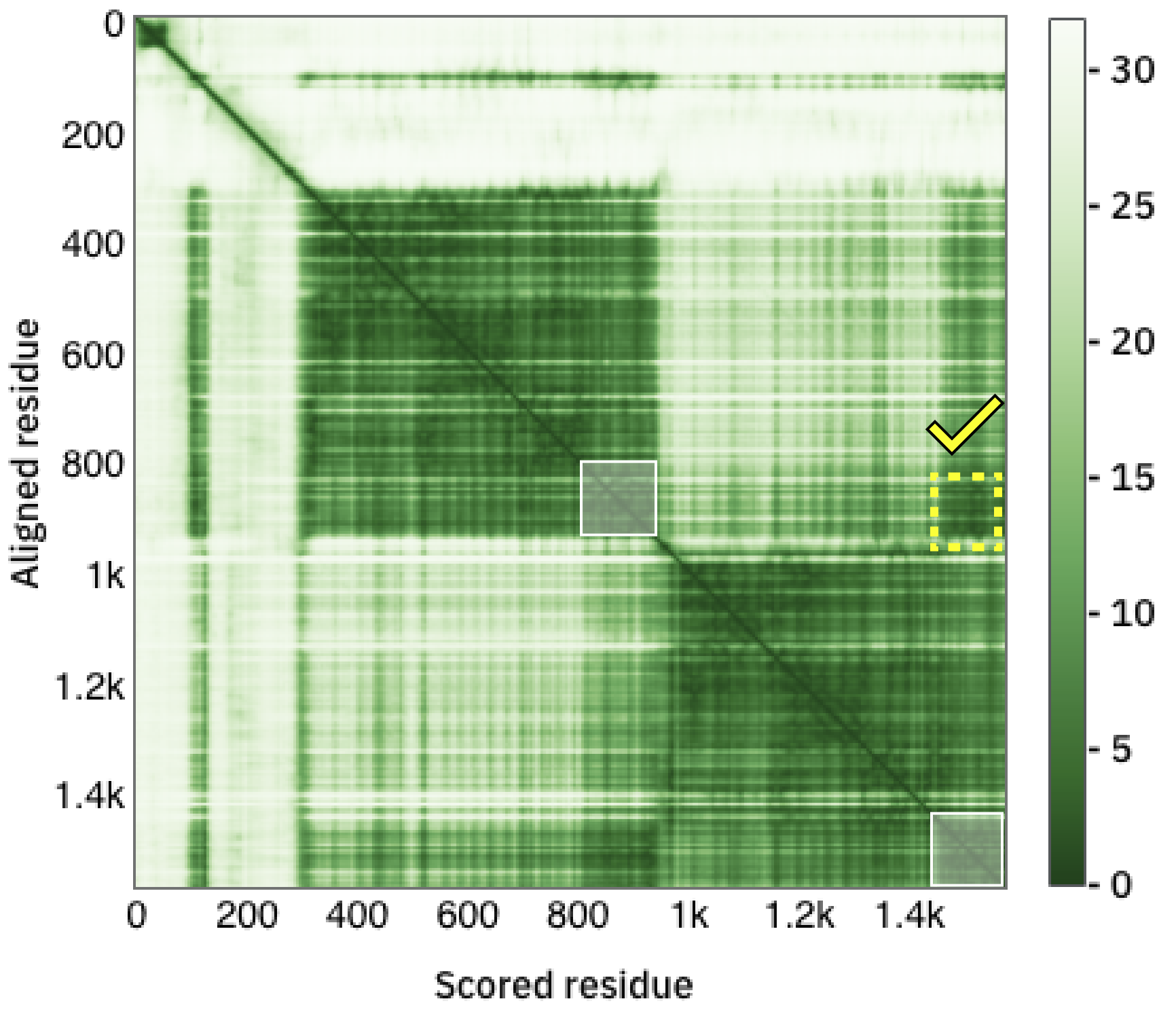
La segunda métrica se denomina PAE (por Predicted Aligned error) y compara el error en la predicción de pares de residuos, esto es el error sobre el residuo y cuando las estructuras real y predicha son alineadas sobre el residuo x. Esta medida permite la identificación global de unidades de plegamiento (dominios) y permite predecir si dos dominios guardan relaciones espaciales definidas, o si tienen variabilidad (por ejemplo, si están conectadas por un linker flexible).
Costo computacional
AlphaFold consume muchísimos recursos. Por lo tanto, muchas proteínas de organismos modelos están siendo modeladas y puestas a disposición de la comunidad científica en una base de datos: https://alphafold.ebi.ac.uk/, como ya pudo observar en UniProt. Por el momento, no se están construyendo modelos de proteínas virales, proteínas grandes y solo se construyeron monómeros (aunque recientemente anunciaron la futura deposición de multímeros).
Si bien el código no fue liberado, AlphaFold fue implementado en un servidor y se puso a disponibilidad de la comunidad.
Ejercicio 2. Predicción estructural de retinoblastoma.
La proteína del retinoblastoma (Rb) es una proteína supresora de tumores que actúa como regulador del ciclo celular en la transición G1-S.
Está formada por un dominio N-terminal, un dominio "pocket", formado por dos subdominios A y B, y un dominio C-terminal.
Al inicio del ciclo, Rb se encuentra unida a los factores de transcripción E2F y remodeladores de la cromatina. A medida que progresa el ciclo Rb es fosforilada y se libera. Permitiendo que se active la transcripción de genes dependientes de E2F necesarios para el progreso del ciclo celular.
Dada las funciones de Rb, es de alta relevancia clínica. Mutaciones en Rb o interacciones con proteínas virales que impidan su interacción con E2F llevan al crecimiento celular y tumores.
- Ingrese al servidor de AlphaFold y copie la secuencia de la proteína Rb. En Entity asegúrese que dice Protein.
>sp|P06400|RB_HUMAN Retinoblastoma-associated protein OS=Homo sapiens OX=9606 GN=RB1 PE=1 SV=2
MPPKTPRKTAATAAAAAAEPPAPPPPPPPEEDPEQDSGPEDLPLVRLEFEETEEPDFTAL
CQKLKIPDHVRERAWLTWEKVSSVDGVLGGYIQKKKELWGICIFIAAVDLDEMSFTFTEL
QKNIEISVHKFFNLLKEIDTSTKVDNAMSRLLKKYDVLFALFSKLERTCELIYLTQPSSS
ISTEINSALVLKVSWITFLLAKGEVLQMEDDLVISFQLMLCVLDYFIKLSPPMLLKEPYK
TAVIPINGSPRTPRRGQNRSARIAKQLENDTRIIEVLCKEHECNIDEVKNVYFKNFIPFM
NSLGLVTSNGLPEVENLSKRYEEIYLKNKDLDARLFLDHDKTLQTDSIDSFETQRTPRKS
NLDEEVNVIPPHTPVRTVMNTIQQLMMILNSASDQPSENLISYFNNCTVNPKESILKRVK
DIGYIFKEKFAKAVGQGCVEIGSQRYKLGVRLYYRVMESMLKSEEERLSIQNFSKLLNDN
IFHMSLLACALEVVMATYSRSTSQNLDSGTDLSFPWILNVLNLKAFDFYKVIESFIKAEG
NLTREMIKHLERCEHRIMESLAWLSDSPLFDLIKQSKDREGPTDHLESACPLNLPLQNNH
TAADMYLSPVRSPKKKGSTTRVNSTANAETQATSAFQTQKPLKSTSLSLFYKKVYRLAYL
RLNTLCERLLSEHPELEHIIWTLFQHTLQNEYELMRDRHLDQIMMCSMYGICKVKNIDLK
FKIIVTAYKDLPHAVQETFKRVLIKEEEYDSIIVFYNSVFMQRLKTNILQYASTRPPTLS
PIPHIPRSPYKFPSSPLRIPGGNIYISPLKSPYKISEGLPTPTKMTPRSRILVSIGESFG
TSEKFQKINQMVCNSDRVLKRSAEGSNPPKPLKKLRFDIEGSDEADGSKHLPGESKFQQK
LAEMTSTRTRMQKQKMNDSMDTSNKEEK
-
Haga click en save job.
-
Ingrese un nombre para su trabajo en el primer campo.
-
Habilite el uso de seed. Y elija un número. Esto permitirá que su trabajo sea reproducible.
-
Verifique que en la parte inferior debe aparecer su trabajo.
-
Haga click en su trabajo y luego en Continue and preview job
-
Haga click en Confirm and submit job
-
El modelo estará listo en unos 5 minutos
-
-
Observe los resultados obtenidos en el servidor,
- ¿Qué puede decir de la estructura de la proteína?
- ¿Cuántos dominios posee? ¿ordenados o desordenados?
- ¿Puede decir aproximadamente los límites?
-
Observe el gráfico de PAE.
- ¿Qué interpreta?
-
Descargue los resultados haciendo click en los tres puntitos y en Download. Se descargará un archivo zip con el nombre que usted utilizó para su trabajo. Descomprima el archivo.
Encontrará 13 archivos que contienen el nombre de su trabajo:
-
*_job-request.jsonTiene los parámetros utilizado en el trabajo.-
ptm: rango 0-1 indicando el valor predicho de TM (template modelling score) para la estructura completa.
-
iptm: rango 0-1 indicando el valor predicho de TM en la interfaz para todas las interfaces en la estructura.
-
fraction_disordered: rango 0-1 indicando que fracción de la estructura predicha es desordenada medido como el area de superficie accesible. (Abramson et al, 2024)
-
has_clash: Booleano (valor yes/no) indicando si la estructura tiene un número significativo de átomos que se superpongan (más del 50% de una cadena o una cadena con más de 100 átomos que se superpongan).
-
ranking_score: rango de -100 a 1.5 que se puede utilizar para clasificar las predicciones. Combina ptm, iptm, fraction_disordered y has_clash en un único número según:
\[ 0.8 \times \mathrm{ipTM} + 0.2 \times \mathrm{pTM} + 0.5 \times \mathrm{disorder} - 100 \times \mathrm{has\_clash} \]-
chain_pair_pae_min: Una matriz \(m x m\) donde m es el número de cadenas. Cada elemento (i,j) contiene el mínimo valor de PAE para las filas restringidas a la cadena i y columnas restringidas a la cadena j.
-
chain_pair_iptm: Una matriz \(m x m\) donde m es el número de cadenas. Cada elemento (i,j) que no está en la diagonal contiene el valor de ipTM restringido a las cadenas i y j. Cada elemento (i,i) en la diagonal contiene el valor de pTM restringido a la cadenas i.
-
chain_ptm: Una lista de largo m, donde m es el número de cadenas. El elemento i contiene el valor de pTM de la cadena i.
-
chain_iptm: Una lista Una lista de largo m, donde m es el número de cadenas. Da el valor promedio de (ipTM) en la interfaz entre cada cadena y todas las otras cadenas.
-
-
*_summary_confidences_0-4.jsonTiene un resumen de las métricas de confianza de los modelos 0 a 4. -
*_full_data_0-4.jsonPara cada uno de los modelos 0 a 4 contiene el PAE, pLDDT, probabilidad de contacto predicha donde contacto se define como una distancia máxima de 8Å (Abramson et al, 2024), entre otros. -
*_model_0-4.cifSon los modelos 0 a 4.
-
-
Abra Chimera y cargue los modelos (si no recuerda File → Open …) y con el mouse seleccione los modelos manteniendo la tecla ctrl presionada para seleccionar más de uno.
-
Alinee los 5 modelos,
- ¿cuál es el RMSD global?
-
Utilice Match Align para ver el alineamiento.
-
¿Qué observa?
-
¿Porque si las secuencias son todas iguales no aparece el n-terminal alineado?
-
-
Los valores de pLDDT están almacenados en la columna del pdb que corresponde a los b-factors. Coloree los modelos según este atributo ingresando en la command line:
rangecolor bfactor 0 orange red 50 white 100 dodger blue- ¿Qué observa?
Ahora cambie los valores para que el programa elija estos valores automaticamente:
rangecolor bfactor min orange red mid white max dodger blueEn el reply log se reportan los valores minimo, medio y máximo encontrados en la columna de b-factors.
- ¿Cuáles son el mínimo y el máximo?
Ahora cambie el valor intermedio (antes puesto automáticamente) a 80 y el máximo a 100:
rangecolor bfactor min orange red 80 white 100 dodger blue- ¿Observa diferencias con lo anterior? ¿Cuáles?
Ahora corra:
rangecolor bfactor 50 orange red 80 white 100 dodger blue- ¿Observa diferencias con lo anterior? ¿Cuáles?
- ¿Porqué considera que elegimos 50 como valor mínimo?
- ¿De qué posición a qué posición consideraría que el modelo es de confianza?
-
Para analizar los valores de pLDDT graficaremos los valores de pLDDT por posición para cada uno de los modelos. Para esto es utilizaremos R Studio y el paquete bio3d que permite leer estructuras de proteínas.
Bio3D no lee correctamente los mmCif generados por AlphaFold3, por lo tanto, vamos a guardas los modelos mmCif en formato
pdb. Para esto, en Chimera con los 5 modelos abiertos vaya a:File → Save PDB...
-
Elija la carpeta donde desea guardar los modelos.
-
En File name elija un nombre para su archivo que incluya $number, por ejemplo,
modelo_rb_$number.Chimera automáticamente va a guardar el modelo 0 en el archivo
modelo_rb_0.pdb, el modelo 1 en el archivomodelo_rb_1.pdbya que reemplaza$numbercon el número de modelo. -
En Save models asegúrese de tener todos los modelos que quiere guardar como pdb seleccionados.
-
En Save multiple models in asegúrese de seleccionar multiple files para tener un archivo por cada modelo.
-
Verifique que en su carpeta se hayan guardado los 5 modelos en formato PDB.
-
Abra R Studio.
# SCRIPT CORREGIDO PARA LEER ARCHIVOS PDB # Las líneas que tienen # al inicio no son parte del script, son comentarios # Cada línea la pueden correr usando ctrl+enter # Instalo el paquete bio3d, esto es necesario correr la primera vez que usamos bio3d install.packages("bio3d") # Cargo el paquete bio3d library(bio3d) # Creo una variable que se llama "directorio" donde voy a guardar la ruta completa a la carpeta # que contiene los modelos, en R si usamos comillas y hacemos tab "busca" la estructura de directorios # y la podemos ir completando hasta llegar a nuestra carpeta deseada. # Esta variable es del tipo "char" o "string" es decir, contiene letras nada más, por eso va entre comillas. directorio <- "/directorio/donde/estan/los/modelos/" # # Creo una variable archivos donde voy a guardar utilizando la función list.files la lista de archivos que están en la carpeta que indiqué en directorio. # Esta variable es un "array" o "vector". Estas variables poseen un índice lo que me permite recorrerlas, por ejemplo: # mi_lista_de_letras <- c("a", "b", "c") # mi_lista_de_letras es una variable de tipo array que contiene en cada posicion un "string". # En la posicion 1 está guardada "a", en la posicion 2 está guarda "b" etc # si imprimo: # mi_lista_de_letras[1] # me devuelve "a" archivos <- list.files(path = directorio,pattern = ".pdb") # si imprimen archivos, les debe devolver la lista de archivos. # para imprimir la variable pueden seleccionarla y apretar: ctrl+enter # Creo la ruta completa para mi archivo 1 usando la funcion paste para "pegar" directorio con lo que está guardado en archivos[1] miarchivo <- paste(directorio,archivos[1],sep="") # Leo el pdb usando la función read.pdb de la librería bio3d mipdb <- read.pdb(file = miarchivo) # Creo una tabla con los datos de interés: el nro de residuo y el valor de plddt almacenado en el campo de b-factors y correspondiendo únicamente a los carbonos alpha. # Para esto creamos la variable "datos" donde guardamos la tabla que en R se llama data.frame # En: mipdb$atom está guarda una tabla que corresponde a la estructura de archivo del PDB. # Pueden imprimirla por consola seleccionandola y haciendo ctrl+enter # En: mipdb$calpha hay valores booleanos, es decir, son TRUE o FALSE. Indican si la línea del PDB que estoy mirando es un Carbono alpha o no. # En: la columna "resno" se guarda el número de residuo # En: la columna "b" se guardan en general los b-factors. En este caso están guardados los valores de pLDDT. datos <- data.frame(Residue = mipdb$atom[mipdb$calpha,"resno"], Rank_1 = mipdb$atom[mipdb$calpha,"b"]) # uso un ciclo for para iterar sobre mi vector donde están guardados los nombres de los archivos y los guardo en distintas columnas de mi tabla datos. Brevemente, para los modelos 2 a 4 voy a ir leyendo los archivos correspondientes y haciendo lo mismo que hicimos para el modelo 1. for (i in 2:length(archivos)){ miarchivo2 <- paste(directorio,archivos[i],sep="") mipdb2 <- read.pdb(miarchivo2) nuevaColumna <- paste("Rank",i,sep="_") datos[nuevaColumna] <- mipdb2$atom[mipdb2$calpha,"b"] } # creo un vector de colores para utilizar en el plot colores <- c("red","blue","purple","green","orange") # creo el nombre del archivo que va a guardar mi plot. # noten que lo va a guardar en el mismo directorio donde están guardados los modelos fileOUT <- paste(directorio,"elijo_nombre_de_archivo.png",sep="") # creo el dispositivo png png(filename = fileOUT, width=20,height=10,units = "cm",res=150) # empiezo un plot plot.new() # ploteo los valores del modelo 1 plot(x = datos$Residue, y = datos$Rank_1, type = "l", xlab = "E7 Residue", ylab = "pLDDT", xlim = c(0,100), ylim = c(0,101), col = colores[1], xaxt='n',yaxt='n') # modifico el EJE X axis(side = 1, at = c(1,seq(5,nrow(datos),by=5)),lwd=0,lwd.ticks = 1) # modifico el EJE Y axis(side = 2, at = seq(5,100,by=5),lwd=0,lwd.ticks = 1) # ploteo los valores de los otros modelos que están guardados en las columnas: 3,4,5,6 for (i in 3:ncol(datos)){ lines(x = datos$Residue, y = datos[,i], col = colores[i-1]) } # creo la leyenda del plot legend(x = 60,y = 40,legend = paste(rep("rank",5),1:5,sep="-"),col = colores,lty = 1,ncol = 2) box(lwd=3) # creo un box en el plot dev.off() # cierro el dispositivo png (si no corren esta línea no van a tener ningún gráfico)-
¿Qué observa?
-
¿Puede identificar las regiones con un pLDDT mayor a 70?
-
¿Puede identificar las regiones con un pLDDT entre 50 a 70?
-
-
Comparando con otras estructuras
-
Abra el pdb: 1n4m.
-
¿Por qué método fue determinada?
-
¿A qué proteína corresponde? ¿De qué organismo?
-
-
Alinee las estructuras utilizando Matchmaker (si no recuerda, Tools → Structure comparison → Matchmaker)
- ¿Cuál es el RMSD global?
Si quieren ver el RMSD por posición sobre la estructura
Structure Comparison → Match align
Seleccione el par de modelos adecuado. En residue-residue distance cutoff seleccione el mismo umbral que utilizó en Matchmaker para pruned atoms (Por defecto es 2)
Para colorear la estructura por RMSD (después de haber hecho Match align) vaya a: Tools → Depiction → Render by Attribute
En attributes of asegúrese que esté seleccionado residues
En el recuadro de Models asegúrese que estén ambos modelos seleccionados.
En la pestaña Render seleccione mavRMSDca y luego haga clic en
Ok. -
Cierre el modelo correspondiente al pdb 1n4m. Via terminal tiene que ingresar el comando close seguido del número del modelo, por ejemplo:
close #0cierra el modelo 0.
O bien, en el model panel, seleccione el modelo correspondiente y haga clic en close.
-
Guarde la sesión y cierre chimera.
-
El algoritmo IUPred será estudiado en mayor profundidad en las próximas clases, por lo tanto, responda brevemete.
IUPred es un predictor de desorden. El score de IUPred varía entre 0 y 1. Un umbral muy utilizado es 0.5. Un score de IUPred mayor a 0.5 se considera una región predicha desordenada o flexible y un score de IUPred menor a 0.5 se considera una región predicha globular u ordenada.
En el gráfico, el eje x son las posiciones de la secuencia y en el eje y el score de IUPred que va de 0 a 1.
Ingrese la secuencia de Rb en IUPRED (actualizada)
- ¿Qué relación encuentra con lo obtenido por Verify3D?
-
En base a los resultados de su análisis, responda:
- ¿Pudo explicar todas las regiones de menor calidad reportadas por Verify3D?
-
Ud. Se sacó un nuevo subsidio donde tiene plata para seguir haciendo estudios estructurales de esta proteína:
- ¿le daría alguna indicación a su nuevo becario, para que tenga más suerte al intentar cristalizarla?